-
Fluoride substitution in LiBH4; destabilization and decomposition
B. Richter, D. Ravnsbæk, M. Sharma, A. Spyratou Stratmann, H. Hagemann and T.R.R. Jensen
Physical Chemistry Chemical Physics, 19 (44) (2017), p30157-30165


DOI:10.1039/C7CP05565J | unige:100150 | Abstract | Article HTML | Article PDF | Supporting Info
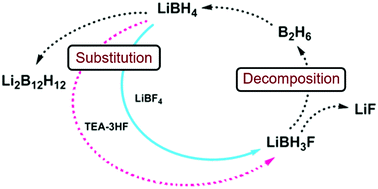
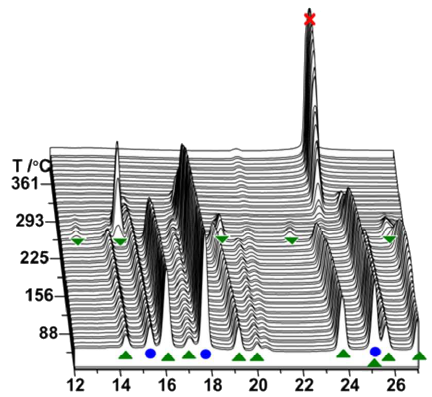
Fluoride substitution in LiBH4 is studied by investigation of LiBH4-LiBF4 mixtures (9:1 and 3:1). Decomposition was followed by in-situ synchrotron radiation X-ray diffraction (in-situ SR-PXD), thermogravimetric analysis and differential scanning calorimetry with gas analysis (TGA/DSC-MS) and in-situ infrared spectroscopy (in-situ FTIR). Upon heating, fluoride substituted LiBH4 forms (LiBH4-xFx) and decomposition occurs, releasing diborane and solid decomposition products. The decomposition temperature is reduced more than fourfold relative to the individual constituents, with decomposition commencing at T / °C = 80 °C. The degree of fluoride substitution is quantified by sequential Rietveld refinement and shows a selective manner of substitution. In-situ FTIR experiments reveal formation of bands originating from LiBH4-xFx. Formation of LiF and observation of diborane release implies that the decomposing materials have a composition that facilitates formation of diborane and LiF, i.e. LiBH4-xFx (LiBH3F). An alternative approach for fluoride substitution was performed, by addition of Et3Nâ3HF to LiBH4, yielding extremely unstable products. Spontaneous decomposition indicates fluoride substitution to have occurred. From our point of view, this is the most significant destabilization effect seen for borohydride materials so far.